

Introduction
Materials and Methods
Materials
Cell culture
Western blotting analysis
Reporter gene assay
2D cellular proliferation assay
3D spheroids growth assay
Spheroids Size Evaluation
Statistical analysis
Results
MAPK signaling regulates ETV4 expressions in ER+ breast cancer cells
Trametinib inhibits ERK phosphorylation and downregulates ETV4 expression
Trametinib suppresses breast cancer cell growth in both 2D and 3D models
ETV4 is transcriptionally upregulated in tamoxifen-resistant breast cancer cells
Activation of MAPK-ETV4 signaling drives tamoxifen resistance and cancer progression
Discussion
Introduction
Estrogen receptor-positive (ER+) breast cancer is the most frequently diagnosed subtype of breast cancers, and metastatic ER+ cancers account for most of the breast cancer-related death (Osborne and Schiff 2011, Zhang et al. 2013). Approximately 70% of human breast cancers are characterized by ER+, establishing endocrine therapy as a pivotal treatment option for ER+ breast cancer (Clark et al. 1984). Endocrine therapies (ET) play a critical role across various stages of the disease, including neoadjuvant, adjuvant, and metastatic settings (Tamoxifen for early breast cancer 1998). ET encompasses various therapeutic options, including a selective estrogen receptor modulator (SERM), including tamoxifen, and aromatase inhibitors (AIs), which are categorized into nonsteroidal agents such as letrozole and anastrozole, and the steroidal agent exemestane, all of which inhibit estrogen production (Tamoxifen for early breast cancer 1998). Tamoxifen (TAM), a well-established SERM, is one of the most frequently prescribed agents in the clinical management of ER+ breast cancer. TAM acts as a competitive antagonist of ER in the ER+ breast tumor, thereby inhibiting estrogen-dependent signaling pathways and suppressing tumor proliferation (Clarke et al. 2003). However, despite its proven efficacy, resistance to tamoxifen remains a significant clinical obstacle. Approximately 30% of patients exhibit intrinsic resistance, while acquired resistance develops in others during treatment, thereby severely compromising the therapeutic benefits of TAM (Jaiyesimi et al. 1995, Tamoxifen for early breast cancer1998, Davies et al. 2011, Zhang et al. 2013). Therefore, identifying novel therapeutic targets for ER+ breast cancer and elucidating the mechanisms underlying acquired tamoxifen resistance represent critical and urgent challenges in the advancement of breast cancer treatment.
ETV4 (ETS variant 4), also known as polyomavirus enhancer activator 3 protein (Pea3), belongs to the E-twenty-six (ETS) transcription factor superfamily, a group of proteins characterized by their conserved ETS DNA-binding domain. Like other members of this family, ETV4 specifically recognizes the GGAA/T core consensus motif, enabling it to regulate the transcription and expression of its target genes (Oh et al. 2012). Recent studies have demonstrated that ETV4 is aberrantly expressed in various human cancers (Hollenhorst et al. 2011, Dumortier et al. 2018, Zhou et al. 2019, Rodriguez et al. 2020, Wang et al. 2020, Xu et al. 2020). Its dysregulation has been shown to play a critical role in driving cancer progression. However, the definitive role of ETV4 and its underlying molecular mechanisms in promoting breast cancer and in acquisition of tamoxifen resistance remains only partially understood, and no clear strategies for targeting ETV4 in therapeutic interventions have been established to date.
Based on the results of this study, we report that trametinib transcriptionally suppresses ETV4 expression through mitogen-activated protein kinase (MAPK) inhibition in ER+ breast cancer cells. We analyzed the growth of ER+ breast cancer both in 2D and 3D cultures to assess breast cancer malignancy. Finally, we measured the ETV4 expression in parental- and TAM-resistant-ER+ breast cancer cells and described the association of ETV4 levels with cancer cell growth. Ultimately, our results indicate effects of trametinib in targeting ETV4 to suppress malignancy in ER+ breast cancer cells.
Materials and Methods
Materials
ETV4 antibody was supplied by Santa Cruz Biotechnology and phospho-ERK (phosphor-p44/42 MAPK) (Thr202/Tyr204), total-ERK, HSP90 antibodies were supplied by Cell Signaling Technology. Trametinib was purchased from MedChemExpress.
Cell culture
MCF-7 cells were cultured at 37°C in 5% CO2/95% atmosphere in DMEM medium (Corning) containing 10% FBS (Gibco), 100 units/mL penicillin, and 100 µg/mL streptomycin (Gibco). TAM-R_MCF-7 cells were established using the method reported elsewhere [200]. Briefly, MCF‐7 cells were washed with PBS, and the culture medium was changed to phenol‐red‐free DMEM containing 10% charcoal‐stripped, steroid‐depleted FBS (Hyclone) and 4‐hydroxytamoxifen (0.1 µM). The cells were continuously exposed to this treatment regimen for 2 weeks and the concentration of 4‐hydroxytamoxifen was gradually increased to 3 µM over a 9‐month period.
Western blotting analysis
After washing with sterile PBS, MCF‐7 or TAMR‐MCF‐7 cells were lysed in RIPA lysis buffer. The cell lysates were centrifuged at 13,000 × g for 10 min to remove the debris, and the proteins were fractionated using a separating gel. The fractionated proteins were then transferred electrophoretically to nitrocellulose membrane, and the proteins were immunoblotted. Horseradish peroxidase‐conjugated anti‐IgG antibodies were used as the secondary antibodies. The nitrocellulose papers were developed using an ECL chemiluminescence system. For ECL chemiluminescence detection, the ChemiDoc Imaging System (Bio-Rad) was used.
Reporter gene assay
Promoter activity was determined using a dual‐luciferase reporter assay system (Promega). Briefly, cells (1 × 105 cells/well) were plated in 12‐well plates overnight and transiently transfected with the LightSwitch promoter reporter plasmid (1 µg) containing ETV4 promoter-RenSP (SwitchGear Genomics) using Fugene6 reagent (Promega). The cells were then incubated in the culture medium with or without trametinib (1 µM) for 24 h, and the Renilla luciferase activities in the cell lysates were measured using a luminometer (Tecan).
2D cellular proliferation assay
For the assessment of cell proliferation in 2D culture, cells were seeded at low density (2 × 103) and were grown for up to 96h. Live cell images were automatically taken 0 h after treatment and then every 24 h by using an CellCyteX System (Cytena) with 10x magnification and the corresponding CellCyteX Software. For analysis of proliferation, phase images were masked to distinguish between background and cells using the basic analyzer provided by the CellCyteX Software. Cell proliferation was determined by analyzing the occupied area over time.
3D spheroids growth assay
3D spheroids were obtained from MCF-7 cells using the method as previously described (Stebbing et al. 2018, Knowlden et al. 2003). Briefly, 100 µL complete medium containing 3 × 103 cells were seeded in each well in an ultra-low attachment 96-well plate (Corning® 96-well Clear Round Bottom Ultra-Low Attachment Microplate). Plates were centrifuged at 300 × g for 3 min and treated with the indicated compounds after 24 h. Treatments were performed adding 100 µL of fresh complete medium with trametinib at the final concentration of 0.001, 0.01, and 0.1 µM into each well. Pictures were taken to assess cell viability every 24 h.
Spheroids Size Evaluation
Spheroids size was evaluated by measuring spheroids perimeter using CellCyteX software (Cytena). Results are expressed as mean pixel measure ± S.D. vs. vehicle-treated control cells from two independent experiments in three replicates.
Statistical analysis
The experiments were performed in at least triplicates. Data were expressed as mean ± S.D. and analyzed by one-way ANOVA (in GraphPad Prism 8), with P-value < 0.05 being considered statistically significant.
Results
MAPK signaling regulates ETV4 expressions in ER+ breast cancer cells
Previous studies indicated that mitogen-activated protein kinase (MAPK)-dependent phosphorylation cascade could enhance ETV4 expression in multiple cancer types (Xiao et al. 2017, Qi et al. 2020). Furthermore, the N-terminus of ETV4 contains conserved MAPK phosphorylation sites, and transactivation is enhanced by the MAPK pathway (de Launoit et al. 1997). The role of the MAPK signaling pathway is well-established across various types of cancers (Degirmenci et al. 2020). Many factors, including receptor tyrosine kinases (RTKs), can activate the MAPK signaling pathway, which plays a pivotal role in tumorigenesis and survival of cancer cells by driving essential biological processes such as proliferation and migration (Degirmenci et al. 2020). To test the role of MAPK signaling in targeting ETV4, we tested trametinib, a MEK1/2 inhibitor (Fig. 1A). Trametinib is an FDA-approved anti-cancer agent that selectively inhibits MEK1/2. Trametinib is primarily used for the treatment of metastatic melanoma associated with BRAF gene mutations (Thota et al. 2015). To assess whether trametinib inhibits ETV4 expression in ER+ breast cancer cells, MCF-7 cells were treated with trametinib at concentrations of 0.001, 0.01 and 0.1 µM (Fig. 1B). Treatment with trametinib led to a reduction in the phosphorylation of ERK, indicating that trametinib effectively inhibits the activity of MEK1/2 responsible for ERK phosphorylation (Fig. 1B). We next investigated the levels of ETV4 expression in MCF-7 cells and found that ETV4 protein expression was significantly reduced by trametinib treatment (Fig. 1C).
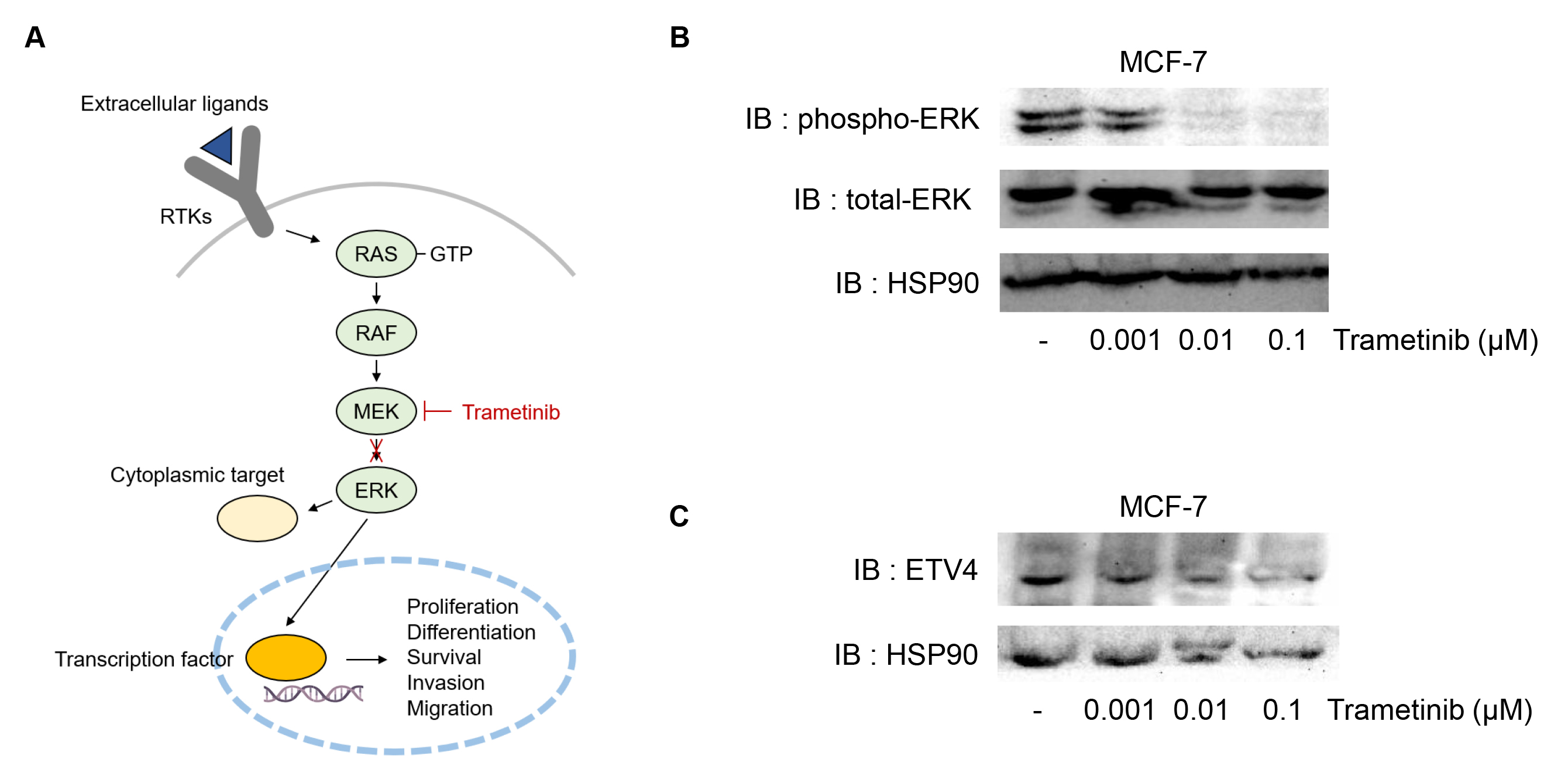
Fig. 1.
Effects of trametinib on ETV4, an effector of MAPK signaling, in MCF-7 cells. (A) Possible mechanism of MAPK activation in ER+ breast cancer. Once extracellular ligands bind to receptor tyrosine kinases (RTKs), the RAS-RAF-MEK-ERK signaling cascade is activated. MEK phosphorylates ERK, which translocated to the nucleus to regulate transcription factors (yellow ovals) involved in cellular processes such as proliferation, differentiation, survival, invasion, and migration. Trametinib, a selective MEK1/2 inhibitor can block the activation of ERK, thereby disrupting downstream signaling and its associated oncogenic effects. (B) Decrease in phosphorylation of ERK by trametinib treatment. The level of phosphorylation of ERK and total level of ERK were determined by western blotting in MCF-7 cells treated with trametinib (0, 0.001, 0.01 or 0.1 µM). (C) Decrease in ETV4 levels by trametinib treatment. MCF-7 cells were treated with trametinib (0, 0.001, 0.01 or 0.1 µM).
Trametinib inhibits ERK phosphorylation and downregulates ETV4 expression
To further explore whether the protein level of ETV4 reduced by trametinib treatment is the results of transcriptional regulation, we examined the effects of transcription factors on ETV4 promoter activity using an ETV4 promoter-RenSP construct (Fig. 2A). In Figure 1, ETV4 expression and ERK phosphorylation were assessed using trametinib at concentrations up to 0.1 µM in the MCF7 cell line. For the measurement of ETV4 promoter activity, three human ER+ breast cancer cell lines (T47D, MCF-7 and BT474) were employed. A concentration of 1 µM trametinib was used across all cell lines, which consistently resulted in a significant reduction in ETV4 promoter activity. In all of the three human ER+ breast cancer cell lines, trametinib treatment decreased the promoter activity of ETV4, indicating that the inhibitory effects of trametinib on ETV4 protein expression could, at least in part, be mediated at the transcriptional level (Fig. 2B-D).
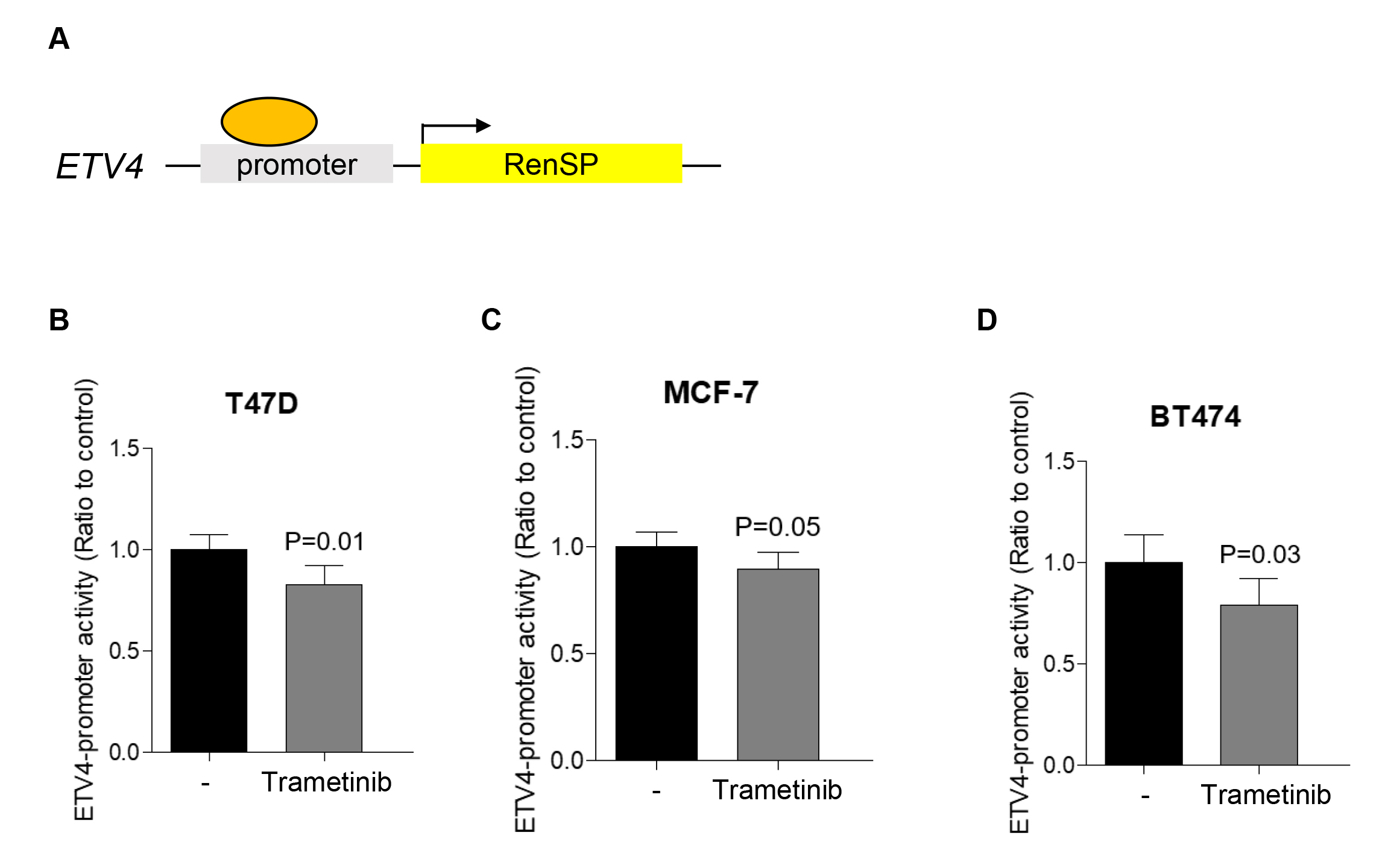
Fig. 2.
Transcriptional regulation of ETV4 by trametinib treatment. (A) Schematic illustration of the RenSP-based reporter construct used in ETV4-promoter assay to examine the transcriptional activity of the ETV4 promoter. Transcriptional activity of ETV4 promoter was determined with or without trametinib treatment (1 µM) in T47D (B), MCF-7 (C), BT474 (D) cells. Data represent means ± S.D.
Trametinib suppresses breast cancer cell growth in both 2D and 3D models
Since growing evidence suggests that ETV4 plays a significant role in promoting cancer cell malignancy (Hollenhorst et al. 2011, Dumortier et al. 2018, Zhou et al. 2019, Rodriguez et al. 2020, Wang et al. 2020, Xu et al. 2020), we tested the effects of trametinib on the 2D growth of MCF-7 breast cancer cells. Data from the 2D growth assay demonstrated that trametinib could suppress MCF-7 cell proliferation in a dose-dependent manner, in comparison with the untreated cells (Fig. 3A). To further test the effects of trametinib on breast cancer growth, we generated and optimized multicellular spheroids from MCF-7 cells in an anchorage-independent 3D environment. 3D spheroids growth assay provides a more physiologically relevant model by mimicking the spatial organization of solid tumors in vivo, enabling more realistic cell-cell mediated signaling compared to 2D cultures. Notably, the growth of MCF-7 spheroids was significantly reduced by trametinib treatment (Fig. 3B). Taking together, these results indicated that the growth-inhibitory effects of trametinib are potent across various conditions, regardless of the distinct cellular cues in 2D and 3D systems, such as cell-cell interactions, cell-ECM interactions, and local gradients of nutrients, growth factors, secreted factors, and oxygen that regulate cell function and behavior (Friedrich et al. 2009, Baker et al. 2012, Lovitt et al. 2013, Lovitt et al. 2014, Wang et al. 2014).
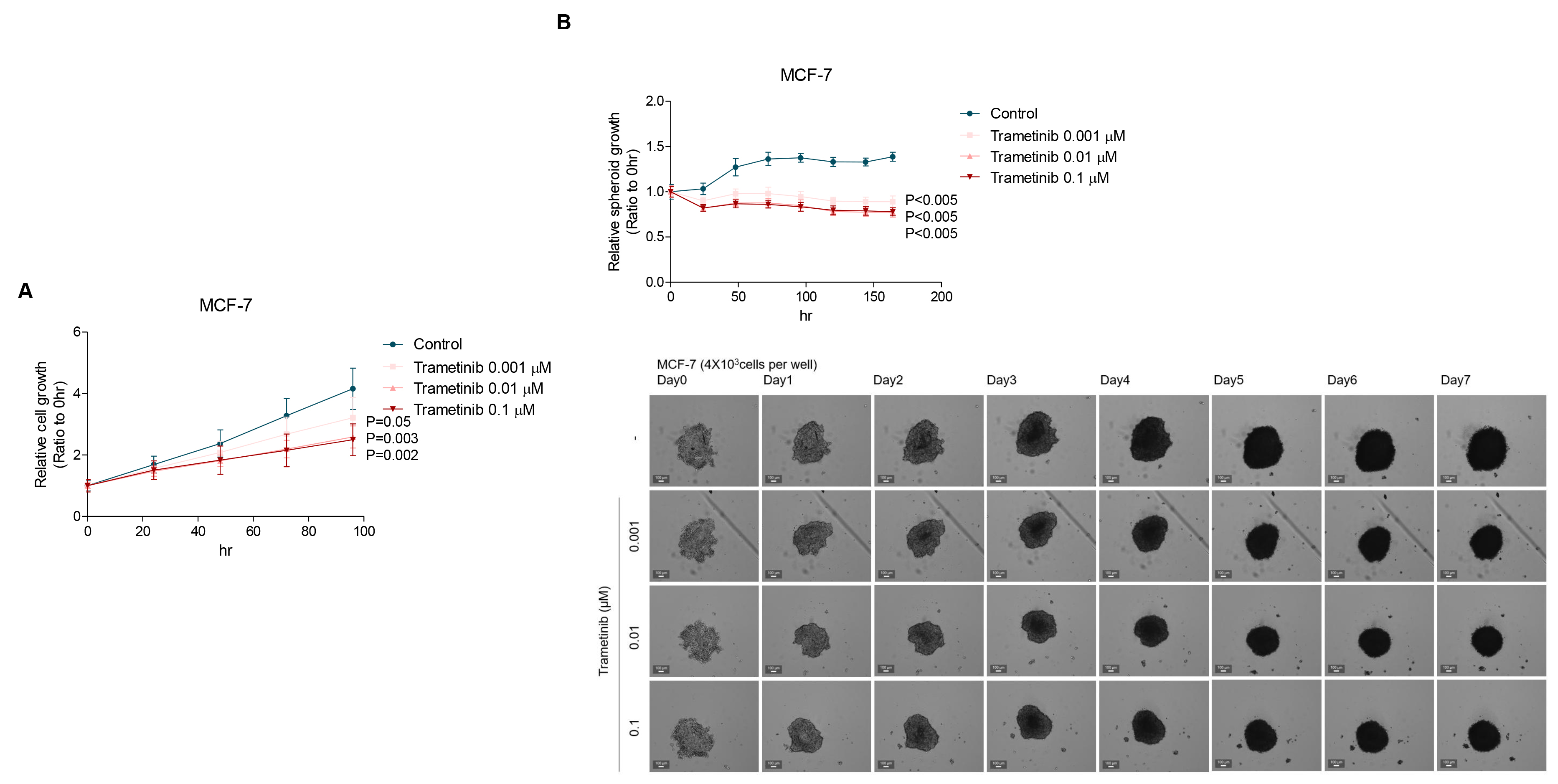
Fig. 3.
2D and 3D growth of MCF-7 cells inhibited by trametinib. (A) Anti-proliferation effect of trametinib on MCF-7 was determined in 2D cultures. MCF-7 cells were treated with 0, 0.001, 0.01, or 0.1 µM trametinib for 96 h. (B) Treatment of MCF-7 3D spheroids with trametinib (0, 0.001, 0.01, or 0.1 µM) for 7 d decreased the growth of spheroids compared with the respective control. Data represent means ± S.D.
ETV4 is transcriptionally upregulated in tamoxifen-resistant breast cancer cells
Tamoxifen (TAM) is the most widely used adjuvant endocrine therapy and has been shown to substantially reduce both recurrence rate and mortality rate (Davies et al. 2011). However, with 5-years use of tamoxifen, one-third of these patients still relapse within 15 years (Davies et al. 2011). Therefore, suppressing the progression of tamoxifen-resistant breast cancer remains an important clinical challenge in ER+ breast cancer. To examine the impact of ETV4 on the TAM-resistant (TAM-R) breast cancer progression, we validated both protein and mRNA levels through western blotting and qRT-PCR. We found that both protein and mRNA levels of ETV4 in TAM-R_MCF-7 cells were increased compared to the parental MCF-7 cells (Fig. 4A-B). These findings suggest that in ER+ breast cancer cells, the acquisition of TAM resistance could be associated with the transcriptional upregulation of ETV4 levels.
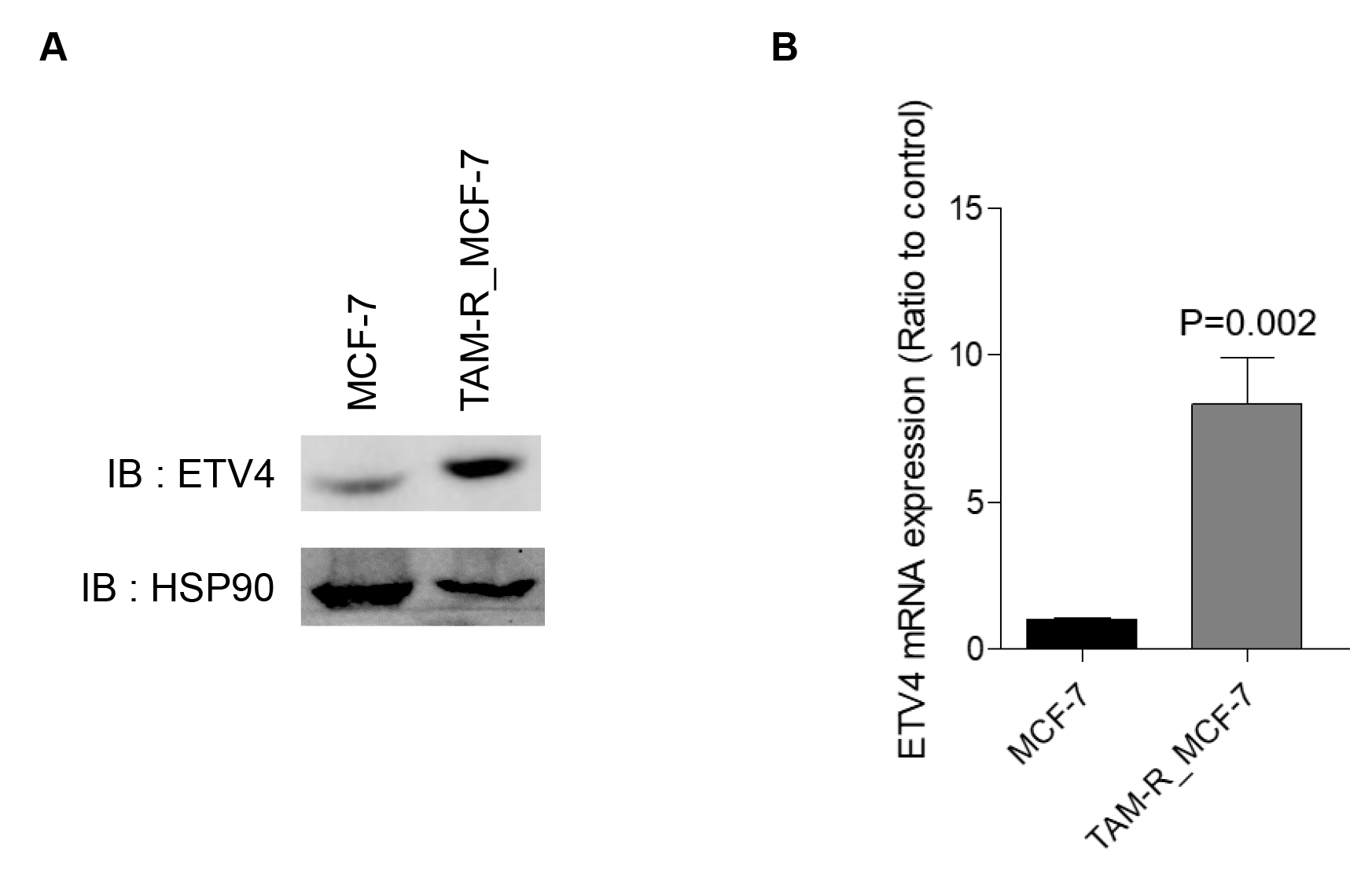
Fig. 4.
Increased ETV4 expression in TAM-R_MCF-7 cells compared to parental MCF-7 cells. (A) A representative immunoblot shows ETV4 protein in both MCF-7 and TAM-R_MCF-7 cells. (B) Real-time qRT-PCR was performed to determine ETV4 mRNA levels in both MCF-7 and TAM-R_MCF-7 cells. Data represent means ± S.D.
Activation of MAPK-ETV4 signaling drives tamoxifen resistance and cancer progression
Since ER-independent proliferation is an essential characteristic of TAM-R cells, we analyzed the proliferation of parental MCF-7 and TAM-R_MCF-7 cells. We observed an increase in proliferation rate in TAM-R_MCF-7 cells compared to parental MCF-7 cells (Fig. 5A). Collectively, these results indicate that as ER+ breast cancer cells progress toward malignancy, activated MAPK signaling may transcriptionally upregulate ETV4 levels, which in turn drive rapid tumor cell growth. As a potential therapeutic approach, we suggest the use of the MEK1/2 inhibitor trametinib to target this pathway.
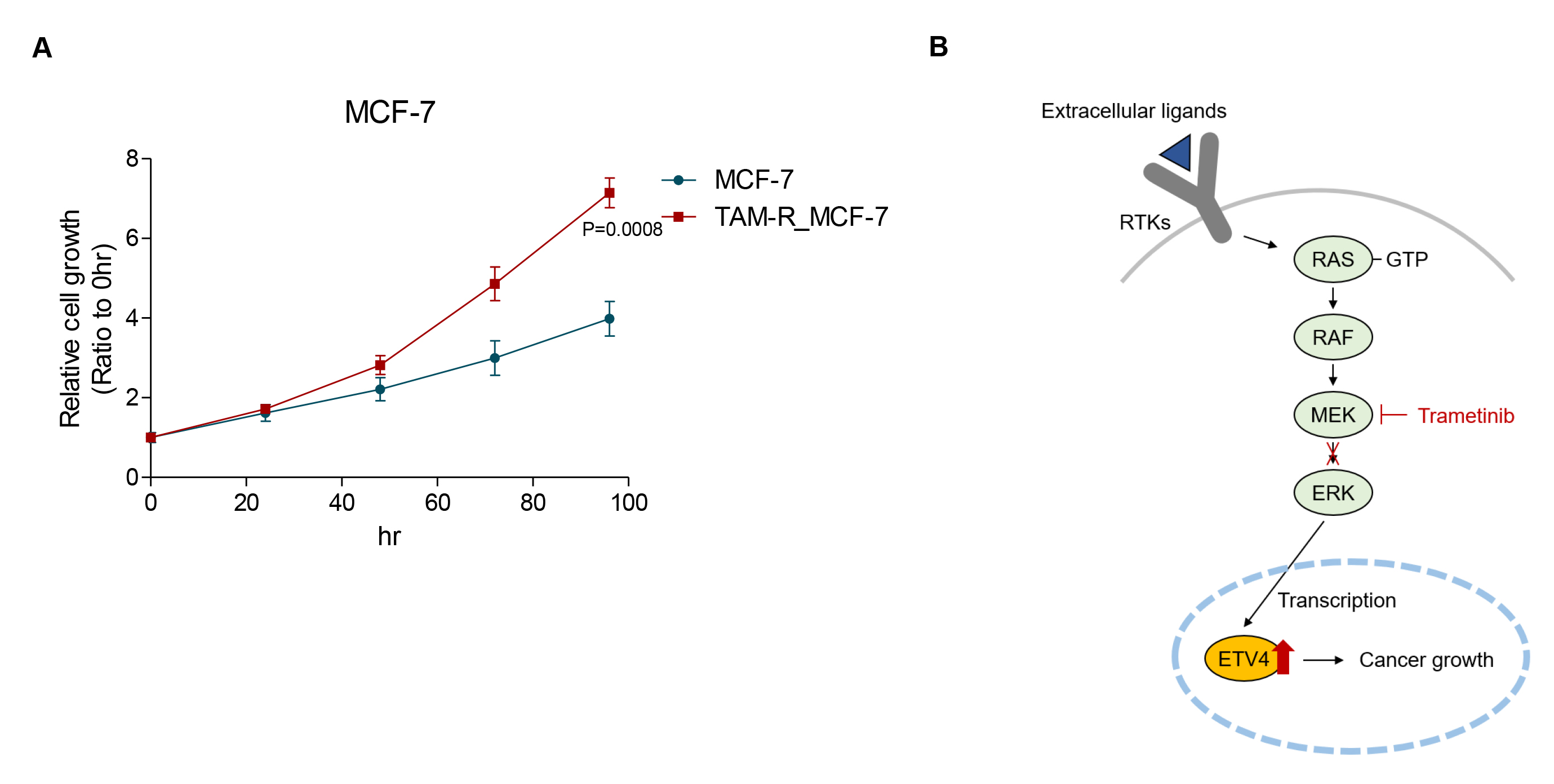
Fig. 5.
Increased cellular proliferation in TAM-R_MCF-7 cells compared to parental MCF-7 cells. (A) Cellular proliferation of both MCF-7 and TAM-R_MCF-7 cells were determined for 96 h. (B) Schematic illustration of trametinib-regulated ETV4 in cancer malignancy, including cell growth. By inhibiting the MAPK signaling pathway, trametinib blocks ERK activation, reducing ETV4-mediated oncogenic processes.
Discussion
Our findings demonstrate that the MAPK signaling pathway plays a critical role in the regulation of ETV4 expression, which contributes to the malignancy of ER+ breast cancer cells. We confirmed that trametinib, a selective MEK1/2 inhibitor, effectively inhibits MEK-dependent ERK phosphorylation, leading to a significant reduction in ETV4 expression at the transcriptional and protein levels. Furthermore, trametinib suppressed MCF-7 breast cancer cell growth in both 2D and 3D culture systems, highlighting its therapeutic potential in targeting tumor proliferation under physiologically relevant conditions mimicking in vivo system. The underlying mechanisms by which MAPK signaling upregulates ETV4 are diverse and multifaceted:
1. ETV4 protein stabilization: ERK activation enhances the stability of the ETV4 protein by preventing its degradation, leading to increased protein levels.
2. Phosphorylation-induced activation: ERK directly phosphorylates ETV4, increasing its transcriptional activity and functional protein levels.
3. Transcriptional upregulation: Activation of the MAPK signaling pathway promotes the transcription of the ETV4 gene, resulting in elevated mRNA and subsequent protein production. Previous studies have identified capicua as a prototypical transcriptional repressor that undergoes degradation upon ERK-mediated phosphorylation, thereby serving as a critical mediator of ERK-driven transcriptional upregulation of ETV4 (Lee et al. 2020).
4. Subcellular localization: ERK-mediated phosphorylation facilitates the nuclear translocation of ETV4, enabling it to function effectively as a transcription factor.
5. Regulation of protein-protein interactions: ERK activation modulates interactions between ETV4 and other proteins, promoting the formation of functional complexes that enhance its activity.
Through these mechanisms, ERK activation collectively increases the protein levels and functional activity of ETV4. In this study, trametinib-mediated MAPK signaling inhibition suppressed ETV4 expression levels, suggesting the involvement of upstream transcription factors regulating ETV4. Further studies need to identify these upstream regulators and elucidate the detailed molecular mechanisms underlying the MAPK-ETV4 axis.
Additionally in TAM-R_MCF-7 breast cancer cells, we observed elevated levels of ETV4, suggesting that tamoxifen resistance might be associated with the transcriptional upregulation of ETV4, driven by MAPK signaling. This upregulation can contribute to ER-independent proliferation, a hallmark of TAM resistance. However, further study is necessary to determine whether trametinib effectively reduces ETV4 levels and suppresses cell growth in TAM-R cells.
Collectively, these results indicate that ETV4 acts as a key driver of tumor progression in ER+ breast cancer cells. The therapeutic efficacy of trametinib in reducing ETV4 expression and suppressing tumor growth supports its potential as a targeted treatment strategy for both tamoxifen-sensitive and resistant breast cancers. These findings highlight the need for further in vivo studies and clinical evaluations to explore the role of trametinib on the broadening of application in overcoming tamoxifen resistance and improving outcomes for patients with ER+ breast cancer.
