

Introduction
Materials and Methods
Animal Treatments
Hepatotoxicity
Lipid Peroxidation
Triglyceride Level in the Liver and Blood
Determination of Hepatic S-Amino Acids and Their Metabolites
Statistical Analysis
Results
Hepatic injury and lipid accumulation
Oxidative stress
Hepatic sulfur amino acids and related metabolites
Discussion
Introduction
Alcohol abuse leads to various organ diseases, with the liver being the most frequently affected organ, a condition known as alcoholic liver disease (ALD) (Namachivayam and Valsala Gopalakrishnan 2021, Wu et al. 2023). Ethanol is metabolized by alcohol dehydrogenase to form acetaldehyde, a toxic substance that can cause liver injury (Namachivayam and Valsala Gopalakrishnan 2021, Wu et al. 2023). Additionally, cytochrome P450 2E1 is another enzyme involved in ethanol metabolism, during which reactive oxygen species are generated, promoting oxidative stress (Namachivayam and Valsala Gopalakrishnan 2021, Wu et al. 2023). Ethanol consumption also induces lipogenesis and impairs lipid export in the liver, resulting in fatty liver (steatosis) (Namachivayam and Valsala Gopalakrishnan 2021). Inflammation triggered by gut-derived lipopolysaccharide activates Kupffer cells, which in turn stimulate stellate cells, leading to the accumulation of collagen fibers, a process known as fibrosis (Namachivayam and Valsala Gopalakrishnan 2021, Wu et al. 2023). Chronic and severe oxidative stress, inflammation, and fibrosis caused by ethanol eventually lead to irreversible cirrhosis and, ultimately, hepatocellular carcinoma (Namachivayam and Valsala Gopalakrishnan 2021, Wu et al. 2023).
S-amino acid metabolism, the metabolic pathway involving methionine and cysteine conversion to taurine and glutathione (GSH), is abundant in the liver, and these substances play critical roles in normal liver function (Jung 2015). Methionine is converted to S-adenosylmethionine (SAM) by methionine adenosyltransferase, and SAM serves as a methyl donor in various methylation reactions (Purohit et al. 2007). One such reaction, catalyzed by phosphatidylethanolamine methyltransferase (PEMT), converts phosphatidylethanolamine (PE) to phosphatidylcholine, a major component of very-low-density lipoproteins (VLDL) (Purohit et al. 2007). Therefore, maintaining SAM concentration and the SAM/SAH ratio are crucial for lipid export from the liver (Purohit et al. 2007). The demethylated product, S-adenosylhomocysteine (SAH), is hydrolyzed to homocysteine, which can induce endoplasmic reticulum stress (Ji and Kaplowitz 2004). Homocysteine can either be recycled back to methionine through methylation or enter the transsulfuration pathway, where it condenses with serine to form cystathionine (Jung 2015). Cystathionine is then cleaved to produce cysteine (Jung 2015). Cysteine serves as a precursor for hypotaurine, which is subsequently oxidized to taurine (Jung 2015). Additionally, GSH, a vital intracellular antioxidant, can be synthesized from cysteine through sequential reactions with glutamate and glycine (Jung 2015).
Previous studies have shown that both acute and chronic ethanol administration can induce fatty liver and liver injury by disrupting S-amino acid metabolism in the liver. In rats fed a Lieber-DeCarli liquid diet containing 5% (w/v) ethanol for six weeks, liver methionine and SAH levels were increased, while SAM, SAM/SAH ratio, cysteine, hypotaurine, taurine, and glutathione (GSH) levels were decreased (Jung et al. 2013). Similarly, in mice, oral ethanol administration (5 g/kg) every 12 hours for a total of three doses elevated liver methionine, hypotaurine, and taurine levels but reduced SAM, cysteine, and GSH concentrations (Kim et al. 2008). Another study using mice treated with ethanol (6 g/kg) twice, 12 hours apart, also demonstrated that acute ethanol administration reduced liver SAM levels and the SAM/SAH ratio, while increasing SAH levels (Arumugam et al. 2022). These findings suggest that both acute and chronic ethanol administration commonly decrease SAM levels and GSH levels, leading to fatty liver development and oxidative stress-mediated liver injury, respectively.
Recent studies have demonstrated that a single oral dose of ethanol administered after 6–12 hours of fasting induces significant liver injury and steatosis in mice. This acute ethanol model has been widely used as an experimental tool to evaluate the hepatoprotective effects of substances such as ZnSO4, Semen Hoveniae extract, Babao Dan capsules, and hydrogen gas (Lambert et al. 2003, Du et al. 2010, Yu et al. 2019, Xu et al. 2024). However, the effects of this single ethanol gavage on hepatic S-amino acid metabolism remain unknown. Therefore, in the present study, we administered a single dose of 5 g/kg ethanol to mice after 12 hours of fasting and analyzed hepatic S-amino acids and related metabolites to investigate the mechanisms underlying ethanol-induced hepatotoxicity and steatosis.
Materials and Methods
Animal Treatments
Male C57BL/6 mice (7 weeks old, n=10) were purchased from Samtako Bio Korea (Osan, Republic of Korea). The animal protocols were approved by the Institutional Animal Care and Use Committee of Jeju National University (2024-0076). After a 1-week acclimation period, the mice were fasted for 12 hours. Normal saline was administered as a vehicle to the control group (n=4), while ethanol (5 g/kg, 50% w/v) was orally administered to the treatment group (n=6). The mice were sacrificed 12 hours after ethanol administration, and blood and liver samples were collected for further analyses.
Hepatotoxicity
Liver injury was assessed by measuring serum alanine transaminase (ALT) activity using an ALT detection kit (Asan Pharm. Co., Ltd., Seoul, Republic of Korea).
Lipid Peroxidation
Thiobarbituric acid-reactive substances (TBARS), the products of lipid peroxidation, were quantified using liver tissue to assess oxidative stress. Liver tissue was homogenized with phosphate-buffered saline (pH 7.4) and reacted with TBA (0.25%), trichloroacetic acid (5%), and HCl (0.17 M) for 40 minutes at 90°C. The centrifuged supernatant was collected, and absorbance was measured at 535 nm.
Triglyceride Level in the Liver and Blood
Lipids in the liver tissue homogenate were extracted using chloroform and methanol. Triglyceride (TG) levels were quantified from the chloroform phase of the centrifuged sample (10,000×g, 10 minutes) or serum using a triglyceride detection kit (Asan Pharm. Co., Ltd., Seoul, Republic of Korea).
Determination of Hepatic S-Amino Acids and Their Metabolites
Hepatic concentrations of methionine, SAM, SAH, homocysteine, serine, cysteine, cystathionine, taurine, hypotaurine, glutamate, and glycine were determined using our previously established HPLC methods (Seo et al. 2023). GSH and oxidized GSH (GSSG) were quantified using a GSH/GSSG-Glo™ Assay Kit (Promega, Madison, WI, USA).
Statistical Analysis
The data are presented as mean ± standard deviation (SD), and statistical differences between groups were calculated using Student’s t-test in Microsoft Excel (Microsoft, Redmond, WA, USA). Statistical significance was defined as a P-value of less than 0.05.
Results
Hepatic injury and lipid accumulation
Acute ethanol administration decreased body (Fig. 1A) and liver weights (Fig. 1B), and their ratio (Fig. 1C). Serum ALT activity indicating liver injury was increased by ethanol treatment (Fig. 1D). The serum TG level remained unaltered (Fig. 1E), but hepatic TG levels were elevated by ethanol treatment (Fig. 1F), as evidenced by the more yellowish appearance of the livers in ethanol-treated mice compared to those of the control mice (Fig. 1G). These results suggest that acute ethanol treatment promoted hepatotoxicity and steatosis in mice.
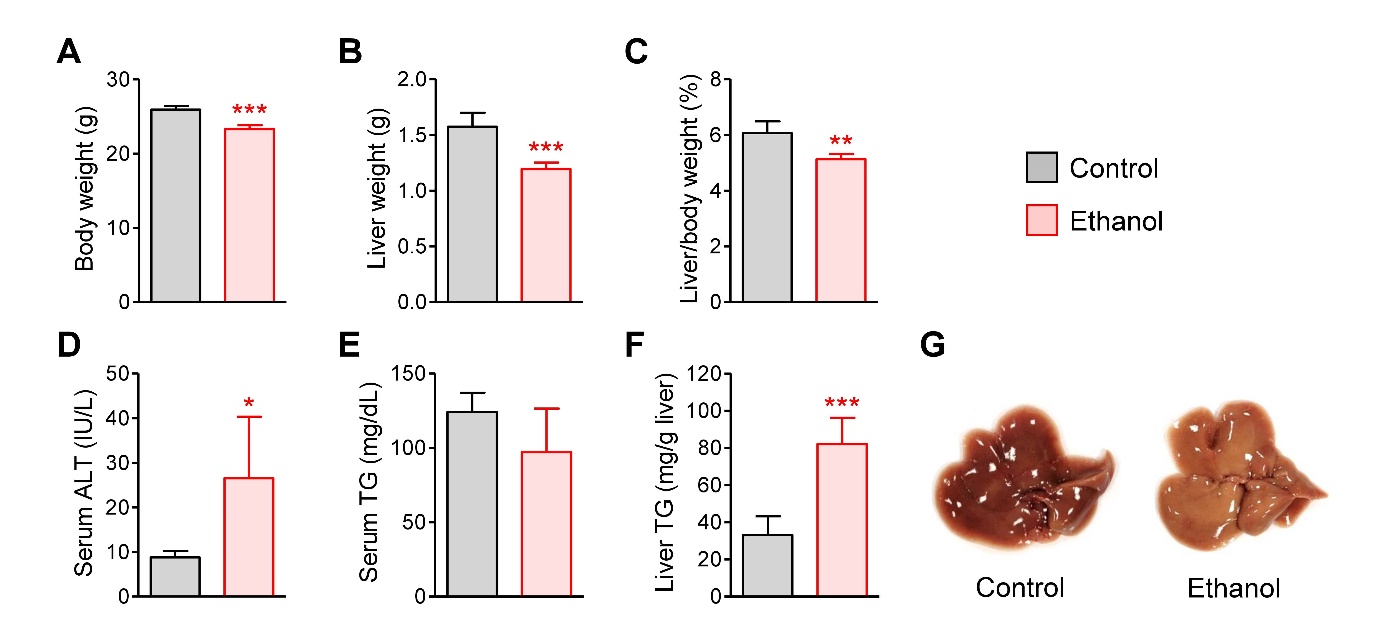
Fig. 1.
Hepatic injury and steatosis induced by acute ethanol administration in mice. (A) Body weight. (B) Liver weight. (C) Liver/body weight ratio (%). (D) Serum alanine aminotransferase (ALT) activity. (E) Serum triglyceride (TG) concentration. (F) Hepatic TG concentration. (G) Liver images from control and ethanol-treated mice. Mice were fasted for 12 h, and administered with normal saline (n=4) or ethanol (5 g/kg, n=6) via oral gavage. After 12 h, liver and blood samples were collected. Student’s t-test, *, **, ***p < 0.05, 0.01, and 0.001, respectively, vs control.
Oxidative stress
Oxidative stress is an important mechanism of acute ethanol-induced hepatotoxicity (Namachivayam and Valsala Gopalakrishnan 2021, Wu et al. 2023). In our study, the hepatic TBARS level, a marker of lipid peroxidation, appeared to be slightly increased by ethanol, but there was no statistical difference between the groups (Fig. 2A). However, the GSH level was significantly decreased by ethanol (Fig. 2B). The GSSG concentration was unchanged (Fig. 2C), but the GSH-to-GSSG ratio, an indicator of cellular redox status, was markedly diminished in the livers of ethanol-treated mice (Fig. 2D).
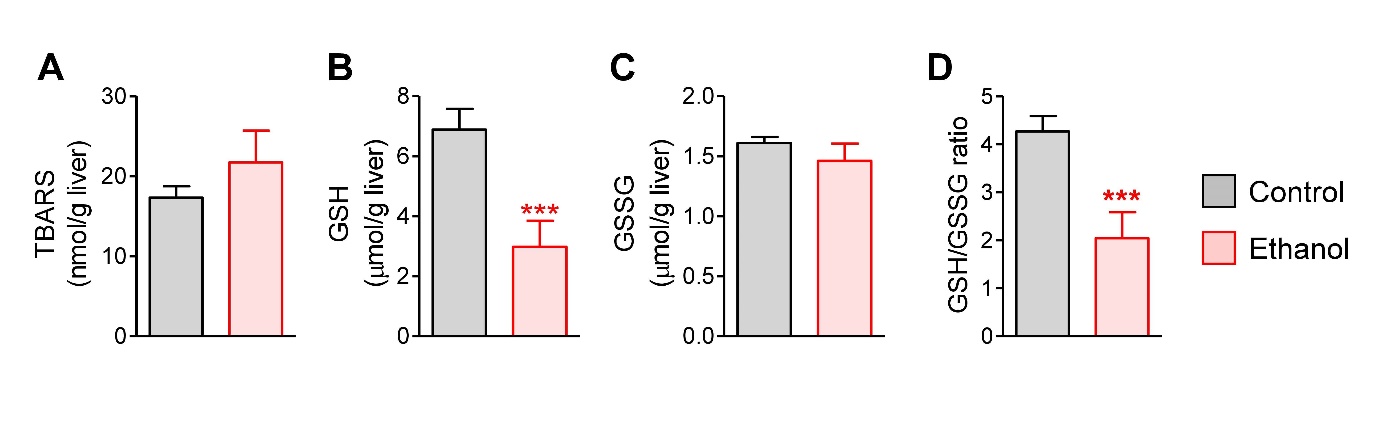
Fig. 2.
Hepatic oxidative stress induced by acute ethanol administration in mice. (A) Hepatic thiobarbituric acid-reactive substances (TBARS) levels. (B) Hepatic glutathione (GSH) level. (C) Hepatic oxidized GSH (GSSG) level. (D) Hepatic GSH/GSSG ratio. Mice were fasted for 12 h, and administered with normal saline (n=4) or ethanol (5 g/kg, n=6) via oral gavage. After 12 h, liver samples were collected. Student’s t-test, ***p < 0.001, vs control.
Hepatic sulfur amino acids and related metabolites
Ethanol induced dramatic changes in hepatic S-amino acid metabolism. Levels of methionine, SAM, SAH, the SAM-to-SAH ratio, hypotaurine, and glutamate remained unchanged (Fig. 3A, 3B, 3C, 3D, 3I, and 3K). However, homocysteine and taurine levels were elevated (Fig. 3E and 3J), while serine, cystathionine, cysteine, glycine, and GSH levels were decreased following ethanol administration (Fig. 3F, 3G, 3H, 3L, and 3M). The increase in homocysteine levels appears to be related to the decrease in serine, resulting in the inhibition of transsulfuration to cystathionine and cysteine, both of which were reduced by ethanol. Although glutamate concentrations were unchanged, the decreased levels of cysteine and glycine suggest that GSH synthesis could be impaired due to the reduction in these two substrates. Furthermore, the increased taurine levels suggest that cysteine may have been diverted toward taurine synthesis rather than GSH synthesis. These changes are summarized in Fig. 3N. The results indicate that acute ethanol administration inhibited the transsulfuration reaction and GSH synthesis, leading to an increase in homocysteine and a decrease in GSH levels in the liver.
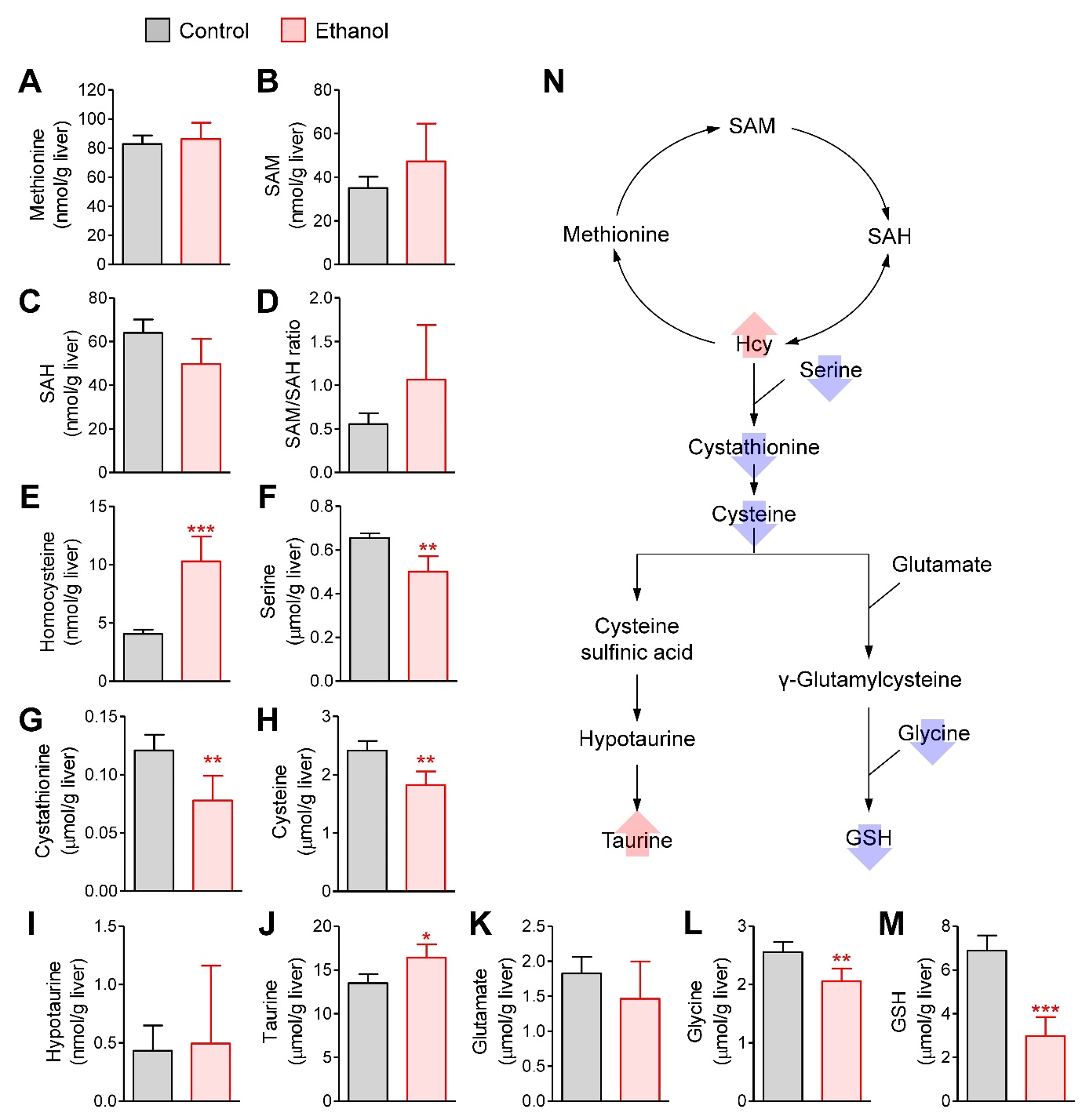
Fig. 3.
Hepatic concentrations of sulfur amino acids and related metabolites. (A) Methionine. (B) S-adenosylmethionine (SAM). (C) S-adenosylhomocysteine (SAH). (D) SAM/SAH ratio. (E) Homocysteine. (F) Serine, (G) Cystathionine. (H) Cysteine. (I) Taurine. (J) Hypotaurine. (K) Glutamate. (L) Glycine. (M) Glutathione (GSH). (N) Summary of changes in S-amino acid metabolism. Mice were fasted for 12 h, and administered with normal saline (n=4) or ethanol (5 g/kg, n=6) via oral gavage. After 12 h, liver samples were collected. Student’s t-test, *, **, ***p < 0.05, 0.01, and 0.001, respectively, vs control.
Discussion
The results of our study show that a single dose of ethanol administered after 12 hours of fasting induces hepatotoxicity and steatosis, as evidenced by increased serum ALT and hepatic TG levels, respectively (Fig. 1). Although lipid peroxidation was unchanged, hepatic GSH concentration and the GSH/GSSG ratio were decreased, indicating oxidative stress and dysregulation of cellular redox status (Fig. 2). These changes are consistent with previous studies using similar fasting and ethanol administration methods in mice (Lambert et al. 2003, Du et al. 2010, Yu et al. 2019, Xu et al. 2024). However, our study demonstrates that this acute ethanol model significantly alters hepatic S-amino acid metabolism, which is closely associated with hepatocellular damage and fatty liver.
In ALD, hepatic substances involved in S-amino acid metabolism are known to undergo significant changes (Mato et al. 2008, Kim et al. 2008, Kharbanda 2009, Jung et al. 2013, Li et al. 2020, Arumugam et al. 2022). Specifically, ethanol reduces SAM concentration and the SAM/SAH ratio, leading to the inhibition of phosphatidylcholine synthesis by PEMT, which in turn suppresses VLDL synthesis (Purohit et al. 2007). Since VLDL is responsible for exporting excessive lipids from the liver, a decrease in VLDL production can play a significant role in hepatic fat accumulation (Purohit et al. 2007). In our study, however, acute ethanol administration did not affect SAM or SAH levels (Fig. 3). Previous reports indicate that ethanol intake increases homocysteine levels (Ji and Kaplowitz 2004), and homocysteine has been shown to inhibit AMP-activated protein kinase-mediated suppression of lipogenesis (Ahn et al. 2016). Therefore, the hepatic fat accumulation observed in ethanol-treated mice (Fig. 1) might be associated with the elevated hepatic homocysteine levels (Fig. 3). Additionally, homocysteine is known to induce endoplasmic reticulum stress by disrupting protein folding, suggesting that the increase in this S-containing substance (Fig. 3) may also contribute to hepatocellular injury caused by acute ethanol exposure (Fig. 1). Cysteine concentration serves as a rate-limiting factor for GSH biosynthesis (Lu 1998). Our results showed that cysteine and glycine levels, but not glutamate levels, were reduced by ethanol treatment (Fig. 3). This reduction in two essential substrates for GSH synthesis likely caused the observed decrease in GSH levels, and the diminished antioxidant capacity may have contributed to oxidative stress-mediated liver injury in ethanol-treated mice.
In conclusion, our results suggest that a single dose of ethanol (5 g/kg) administered to 12-hour fasted mice induces liver injury and steatosis, which may be associated with alterations in hepatic S-amino acid metabolism. Decreased transsulfuration and impaired GSH synthesis appear to contribute to hepatic steatosis and cellular damage caused by ethanol administration. However, the molecular link between these metabolic changes and the development of ALD remains unclear, necessitating further studies to elucidate the underlying mechanisms. Despite this limitation, our study’s acute ethanol administration protocol may serve as a useful model for investigating metabolic dysfunction in ALD, complementing other acute ethanol rodent models.
